5. Mutation calling
Nextclade calls nucleotide and aminoacid mutations relative to multiple targets.
Mutations relative to reference sequence
In order to detect nucleotide mutations, aligned nucleotide sequences are compared with the reference nucleotide sequence, one nucleotide at a time. Mismatches between the query and reference sequences are then noted and reported differently, depending on their nature:
Nucleotide substitutions: a change from one character to another. For example a change from
Ain the reference sequence toGin the query sequence. They are shown in sequence views in Nextclade Web as colored markers, where color signifies the resulting character (in query sequence).Nucleotide deletions (“gaps”): nucleotide was present in the reference sequence, but is not present in the query sequence. These are indicated by the “
-” character in the alignment sequence. They are shown in sequence views in Nextclade Web as dark-grey markers. In the output files deletions are represented as numeric ranges, signifying the start and end of the deleted fragment (for example:21765-21770)Nucleotide insertions: additional nucleotides in the query sequence that were not present in the reference sequence. They are stripped from the alignment and reported separately, showing the position in the reference after which the insertion occurred and the fragment that was inserted.
22030:ACTwould indicate that the query sequence has the three basesACTinserted between position22030and22031in the reference sequence (the indices are 1-based).
Nextclade also gathers and reports other useful statistics, such as the number of contiguous ranges of N (missing) and non-ACGTN (ambiguous) nucleotides, as well as the total counts of substituted, deleted, missing and ambiguous nucleotides. You can find this information in the results table of Nextclade Web and in the output files of Nextclade CLI.
Similarly, aminoacid mutations and statistics are gathered from the aligned peptides obtained after translation. This step only runs if a genome annotation is provided.
Private mutations
Following the tree placement, Nextclade identifies “private mutations” - the mutations between the query sequence and the sequence corresponding to the nearest neighbor (parent) on the tree.
In the figure, the query sequence (dashed) is compared to all sequences (including internal nodes) of the reference tree to identify the nearest neighbor. The yellow and dark green mutations are private mutations, as they occur in addition to the 3 mutations of the attachment node.
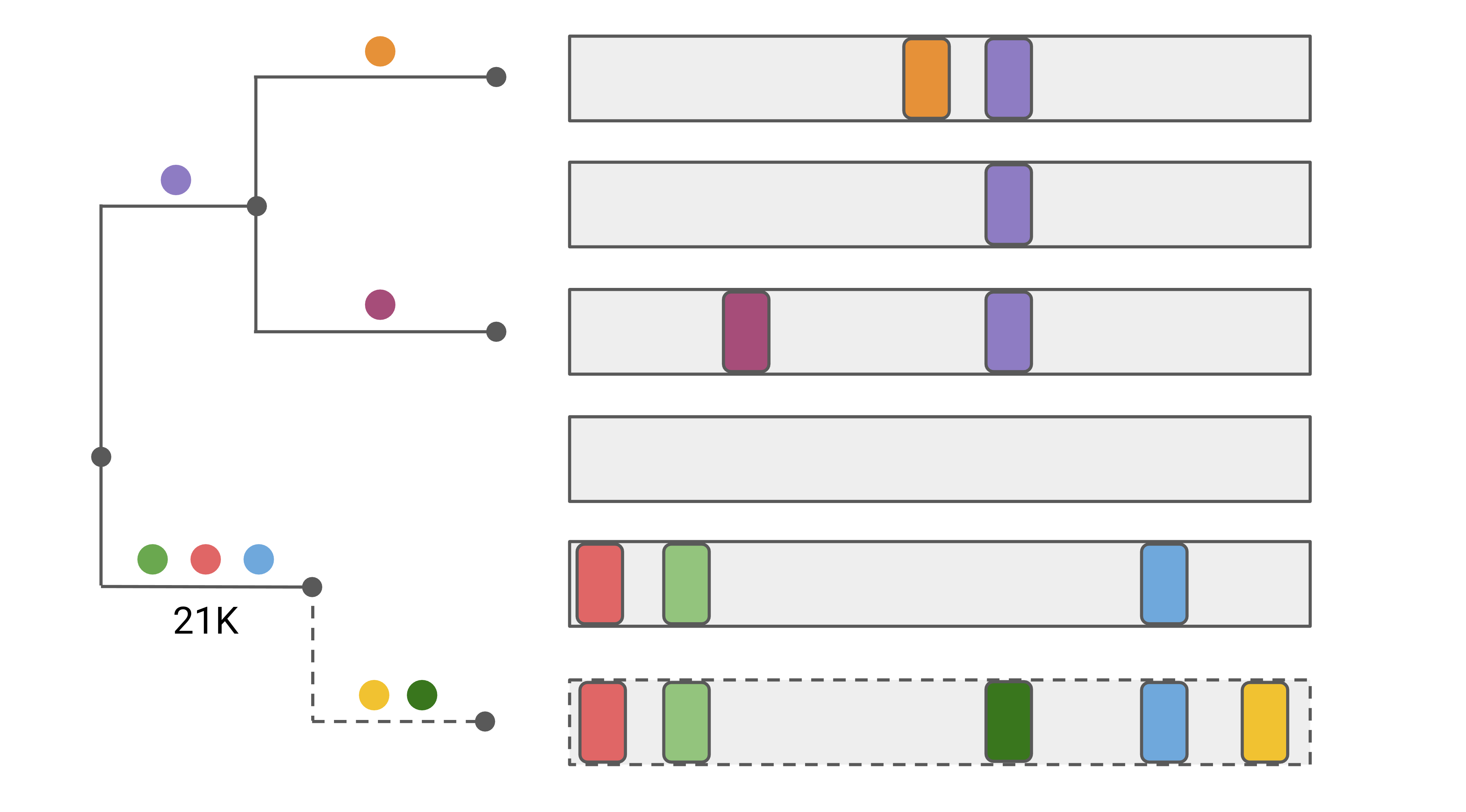
Many sequence quality problems are identifiable by the presence of private mutations. Sequences with unusually many private mutations are unlikely to be biological and are thus flagged as bad.
Nextclade classifies private mutations further into 3 categories to be more sensitive to potential contamination, co-infection and recombination:
Reversions: Private mutations that go back to the reference sequence, i.e. a mutation with respect to reference is present on the attachment node but not on the query sequence.
Labeled mutations: Private mutations to a genotype that is known to be common in a clade.
Unlabeled mutations: Private mutations that are neither reversions nor labeled.
For an illustration of these 3 types, see the figure below.
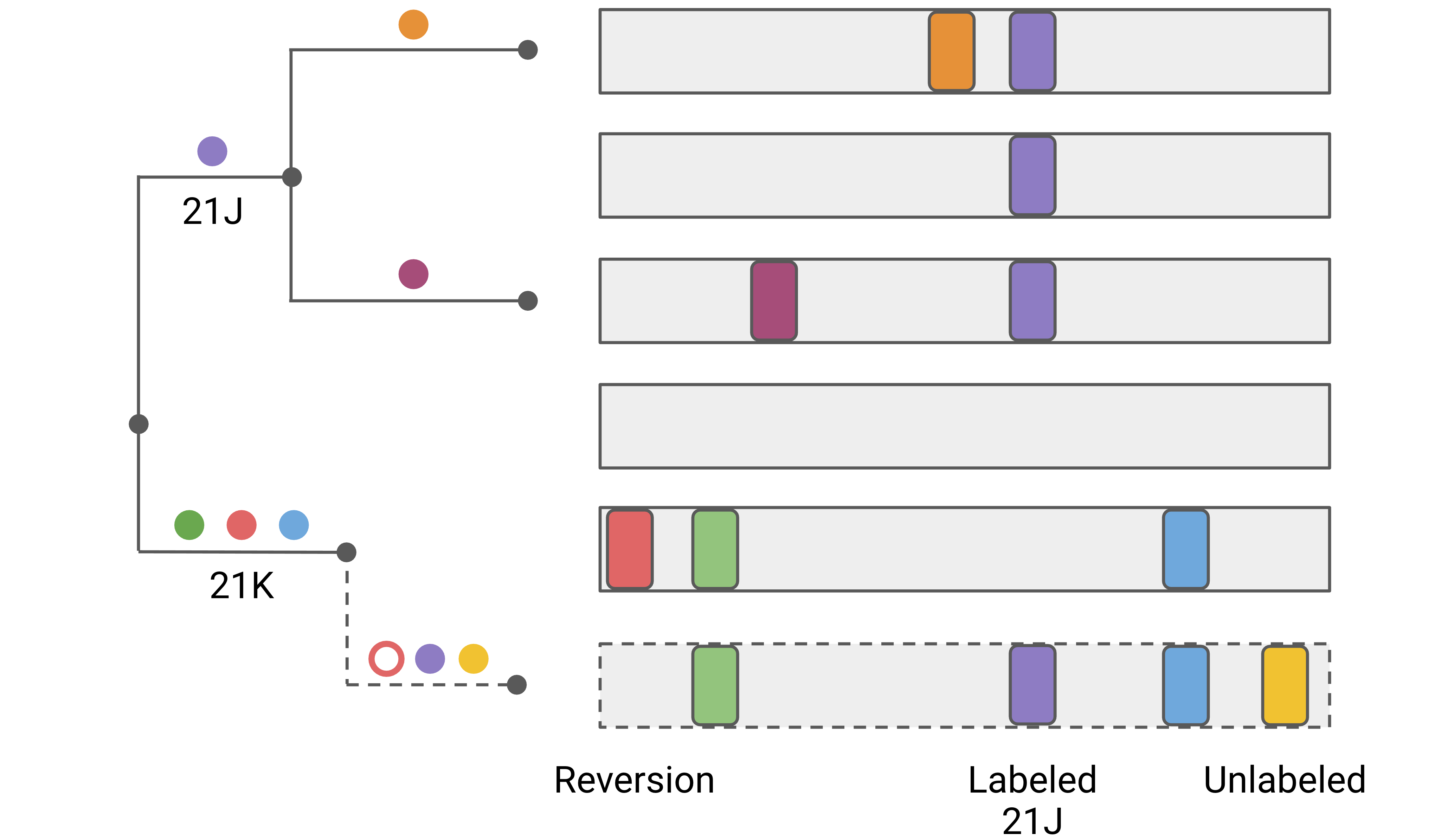
Reversions are common artefacts in some bioinformatic pipelines when there is amplicon dropout and missing sequence is “fill-in” with the reference. They are also a sign of contamination, co-infection or recombination. Labeled mutations are also a common sign of contamination, co-infection or recombination and deserve special attention.
For some datasets, reversions and labeled mutations are therefore weighted several times higher than unlabeled mutations due to their higher sensitivity and specificity for quality problems (and recombination).
In February 2022, the SARS-CoV-2 dataset weighed every reversion 6 (weightReversionSubstitutions) while every labeled mutation was weighed 4 times (weightLabeledSubstitutions). Unlabeled mutations get weight 1 (weightUnlabeledSubstitutions).
From the weighted sum, 8 (typical) is subtracted. The score is then a linear interpolation between 0 and 100 (and above), where 100 corresponds to 24 (cutoff).
Private deletion ranges (including reversion) are currently counted as a single unlabeled substitution, but this could change in the future.
Clade founder search and mutations relative to clade founder
For each query sample possessing a clade, Nextclade finds a corresponding “clade founder” node in the reference tree - the most ancestral node having the same clade. It starts with parent node (nearest node) obtained during tree placement and traverses the tree towards the root, until it finds the last node with the same clade as the parent node.
After that Nextclade calls nucleotide and aminoacid mutations relative to the clade founder.
The search and mutation calling happens separately for clades as well as for each custom clade-like attribute (unless excluded in the pathogen config).
Clade founder search is a built-in convenience wrapper for a node search and relative mutations with pre-agreed search criteria (matching clades).
⚠️ Nextclade assumes that all clades and clade-like attributes defined on the input reference tree are monophyletic. In this context it means that that all nodes belonging to one clade are a single connected component on the tree. Moreover, tree should be sufficiently large and diverse, such that early samples of each of the clades are well represented. Nextclade official datasets enforce these requirements, however third-party dataset authors and users of their datasets need to take additional care.
Arbitrary node search and relative mutations
In addition to the built-in search for clade founder nodes (see above), dataset authors may define criteria for arbitrary nodes of interest on the reference tree. Nextclade will then search these nodes, similarly to how it finds clade founder nodes, and will calculate mutations relative to each of these nodes.
This could be useful, for example, for comparing sequences to the vaccine strains.
Results
The mutation calling step results in a set of mutations and various practical metrics for each sequence.
Mutations can be viewed in the last column of the results table in Nextclade Web.
The “Genetic feature” dropdown allows switching between nucleotide sequence and CDSes (if genome annotation is provided). The “Relative to” dropdown allows to select the target for comparison:
“Reference” - shows mutations relative to the reference sequence
“Parent” - shows private mutations, i.e. mutations relative to the parent (nearest) node
“Clade founder” - shows mutations relative to clade founder
“
founder” - shows mutations relative to clade-like attribute founder (if any defined) any additional entries show mutations relative to the node(s) found according to the custom search criteria (if any defined)
The “Mut” column shows total number of nucleotide mutations and its mouseover tooltip lists the mutations.
All results are emitted into the output JSON, CSV and TSV files in Nextclade CLI and in the “Export” dialog of Nextclade Web.