How to interpret the phylogenetic trees
Transmission trees vs phylogenetic trees
Pathogens spread through rapid replication in one host followed by transmission to another host. An epidemic can only take off when one infection results in more than one subsequent infections.
As the pathogen replicates and spreads, its genome needs to be replicated many times and random mutations (copying mistakes) will accumulate in the genome. Such random mutations can help to track the spread of the pathogen and learn about its transmission routes and dynamics.
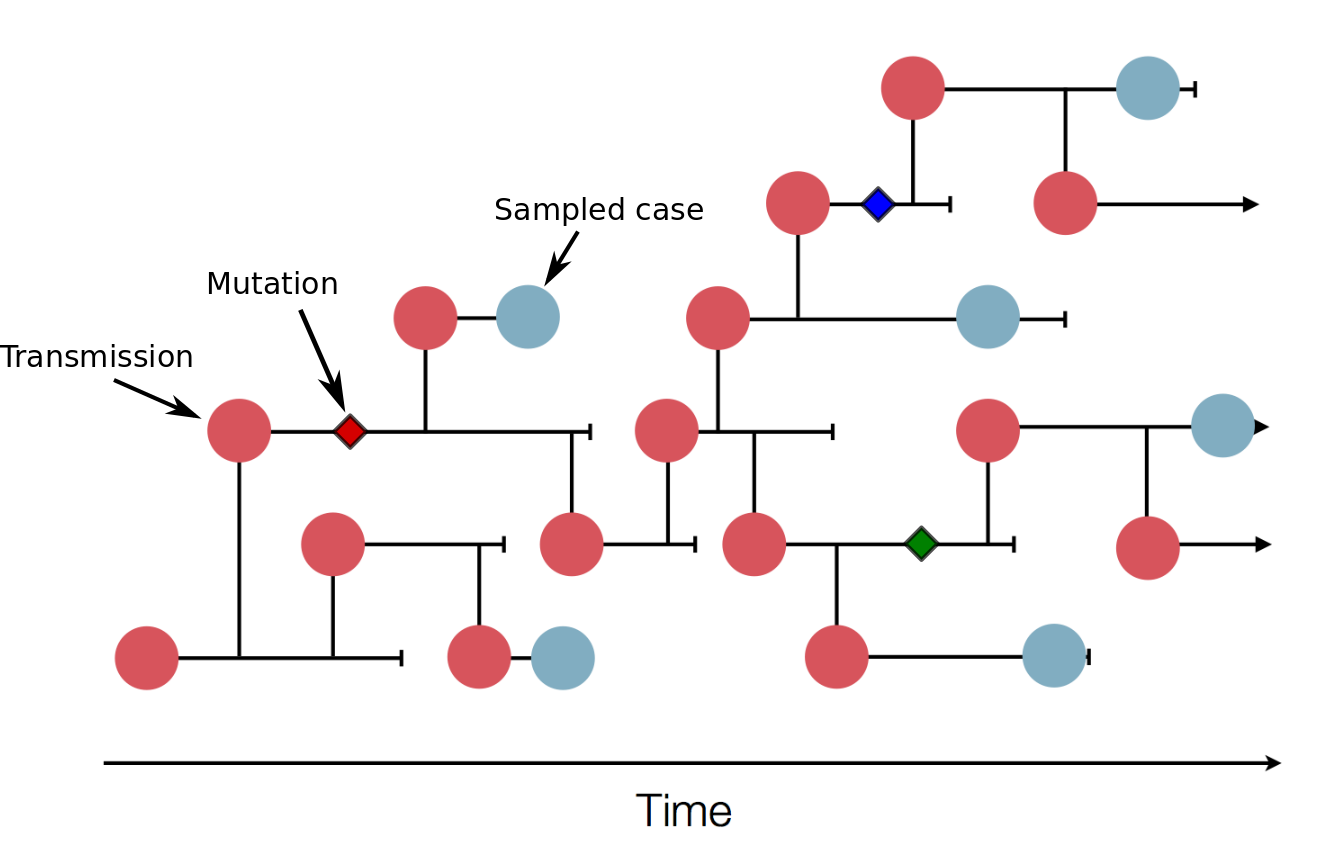
The illustration above shows a sketch of a transmission tree with a subset of cases that were sampled (blue). In practice, the transmission tree is unknown and typically only rough estimates of case counts are available. Genome sequences allow us to infer parts of the transmission tree. In this example, three mutations (little diamonds) are indicated on the tree. Sequences that have the same mutations are more closely related, so these mutations allow us to group samples into clusters of closely related viruses that belong to the same transmission chains.
Reading a Phylogenetic Tree
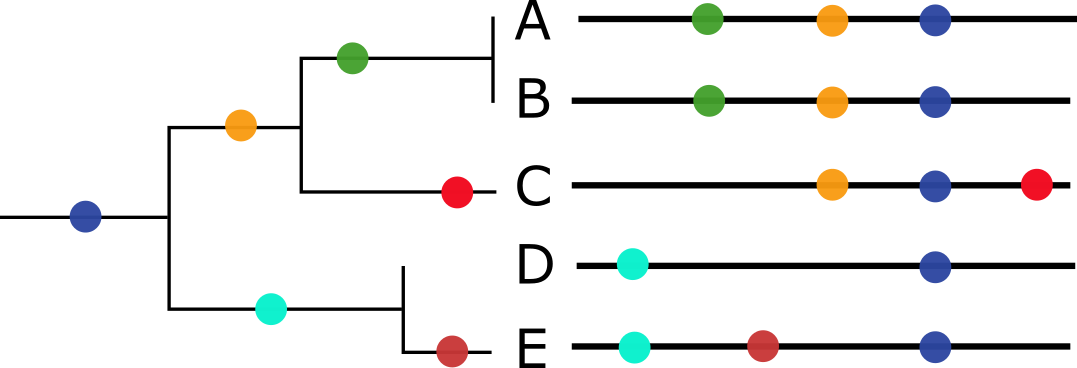
Below, we see an illustration with a phylogenetic tree on the left, where mutations are shown as colored circles. On the right are the corresponding sequences, also with mutations shown as colored circles. We can see that sequences that share the same mutations group together. When sequences appear linked by a flat vertical line, like A and B, this means there are no differences between them – their sequences are identical.
When a sequence sits on a long line on its own, like C or E, this means it has unique mutations not found in other sequences. The longer the line, the more mutations. A and B also have unique mutations (the green circle) not shared by the other sequences, but they are identical to each other.
At the moment, the novel coronavirus (COVID-19) phylogeny may not look much like a ‘tree’. Many of the sequences are identical – they sit together on vertical lines like A and B (some are on the left-most part of the tree). Others have unique or shared mutations and so sit on lines, or ‘branches’, going to the right. You can see how many mutations a branch has by hovering your mouse over it.
Reading a typed Phylogenetic Tree
Phylogenetic trees often contain additional information, such as where geographically individual sequences were isolated from. Additionally, possible locations of internal nodes can be inferred using mathematical models as well. Interpreting these should, however, be done with caution, as the sampling and sequencing or lack thereof can significantly influence the interpretation,
In the following example, we first show fully sampled phylogenetic tree, with samples from two different locations denoted by orange and blue. In the fully sampled case on the left, our interpretation of what happened, was that there were three different introductions from orange to blue. When removing the one orange sequence in the middle, our interpretation is now that there was one introduction into blue that happened much earlier. In the last example, we have only one sequence from orange, which could lead us to think that there was one introduction from blue into orange.
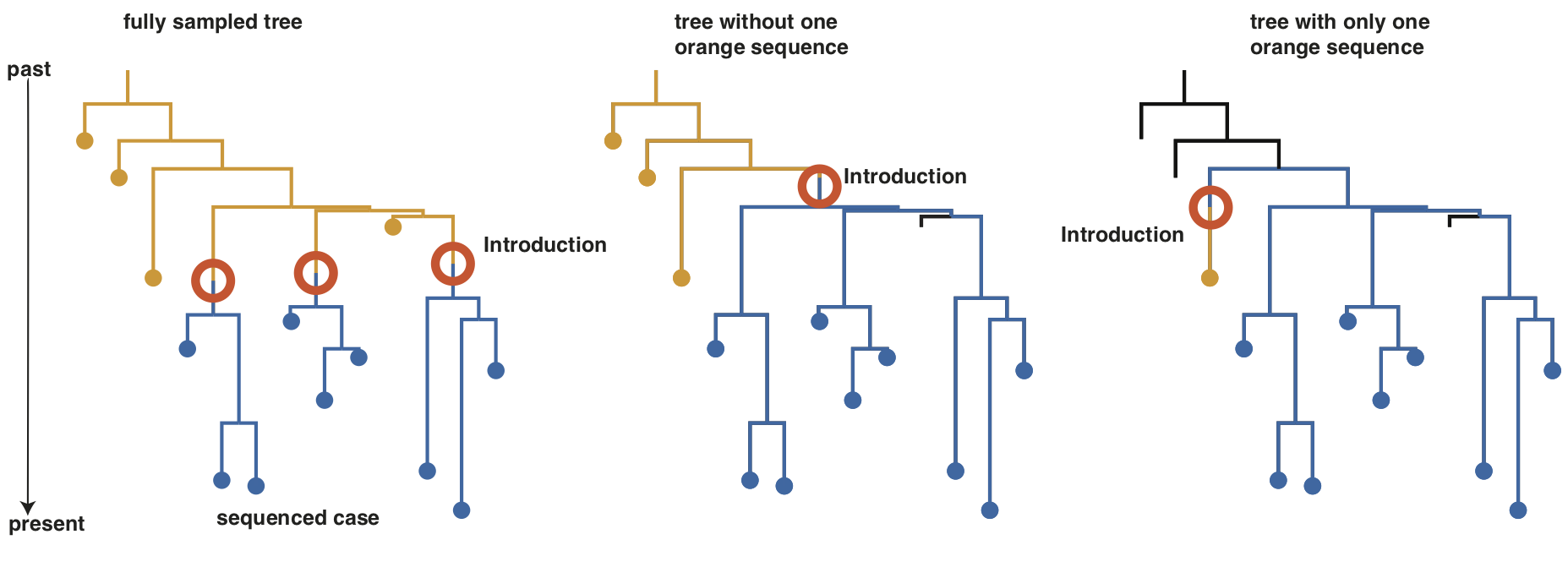
Overall, the inferred locations of where a lineage has been in the past, should be considered as highly uncertain.