Nextstrain is made up of many different parts that all work together. Two core
parts are Augur and Auspice.
Augur is a series of composable, modular bioinformatics tools. We use these
to create recipes for different pathogens and different analyses, which can be
reproduced given the same input data and replicated when new data is available.
Auspice is a web-based visualization program, to present and interact with
phylogenomic and phylogeographic data. Auspice is what you see when, for
example, you visit nextstrain.org/mumps/na.
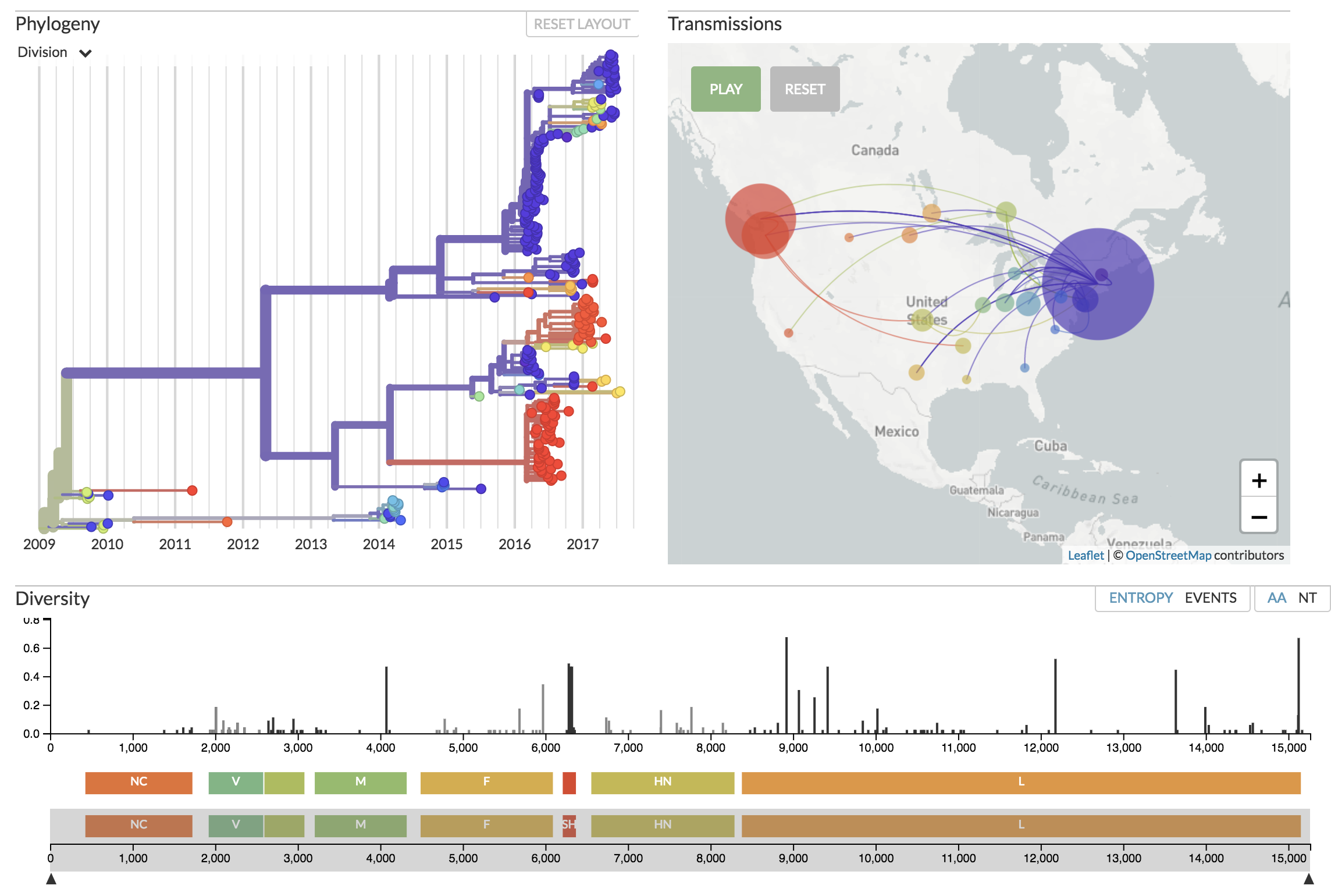
Auspice displaying Mumps genomes from North America.
Datasets are produced by Augur and
visualized by Auspice. These files are often referred to as JSONs
colloquially because they use a generic data format called JSON.
A build is a recipe of several commands and data that produce a single dataset.
nextstrain.org is a web application to host and
present the core pathogen datasets maintained by the Nextstrain team, as well as
datasets published to Nextstrain Groups
and community pages which are
maintained and shared by many other people. The
website incorporates a customized version of Auspice for displaying each
dataset.
You can run Augur and Auspice on
your own computer and use them independently or together with your own builds,
our core builds, or others’ group or community builds. You can even install
Auspice on your own web server if you don’t want
to host your datasets via nextstrain.org.
The Nextstrain CLI ties
together all of the above to provide a consistent way to run pathogen workflows,
access Nextstrain tools like Augur and Auspice across computing environments
such as Docker, Conda, and AWS Batch, and publish datasets to nextstrain.org.
Nextclade is a web application and a command-line
tool for performing viral genome alignment, mutation calling, clade assignment,
quality checks, and phylogenetic placement. Nextclade can be used independently
of other Nextstrain tools as well as integrated into workflows.