Run using example data
This first tutorial introduces our SARS-CoV-2 workflow. You will run the workflow using a small set of reference data that we provide. Subsequent tutorials present more complex scenarios that build on this approach.
Prerequisites
Setup and installation. These instructions will install all of the software you need to complete this tutorial and others.
Setup
Change directory to the
ncovdirectory:cd ncov
Download the example tutorial repository into a new subdirectory of
ncov/calledncov-tutorial/:git clone https://github.com/nextstrain/ncov-tutorial
Run the workflow
From within the ncov/ directory, run the workflow using a configuration file provided in the tutorial directory:
nextstrain build . --configfile ncov-tutorial/example-data.yaml
Break down the command
The workflow can take several minutes to run. While it is running, you can learn about the parts of this command:
nextstrain build .This tells the Nextstrain CLI to build the workflow from
., the current directory. All subsequent command-line arguments are passed to the workflow manager, Snakemake.
--configfile ncov-tutorial/example-data.yaml--configfileis a Snakemake option used to configure the ncov workflow. It takes a file path as the value.ncov-tutorial/example-data.yamlis the value given to--configfile. It is a config file that provides custom workflow configuration including inputs and outputs. The contents of this file with comments excluded are:inputs: - name: reference_data metadata: https://data.nextstrain.org/files/ncov/open/reference/metadata.tsv.xz sequences: https://data.nextstrain.org/files/ncov/open/reference/sequences.fasta.xz refine: root: "Wuhan-Hu-1/2019"
The
inputsentry provides the workflow with one input namedreference_data. The metadata and sequence files refer to a sample of approximately 300 sequences maintained by the Nextstrain team that represent all Nextstrain clades annotated for SARS-CoV-2. The workflow downloads these files directly from the associated URLs. See the complete list of SARS-CoV-2 datasets we provide through data.nextstrain.org.The
refineentry specifies the root sequence for the example GenBank data.For more information, see the workflow configuration file reference.
Running the workflow produces two new directories:
auspice/contains a few files that represent a Nextstrain dataset to be visualized in the following section.results/contains intermediate files generated during workflow execution.
Visualize the results
Run this command to start the Auspice server, providing auspice/ as the directory containing output dataset files:
nextstrain view auspice/
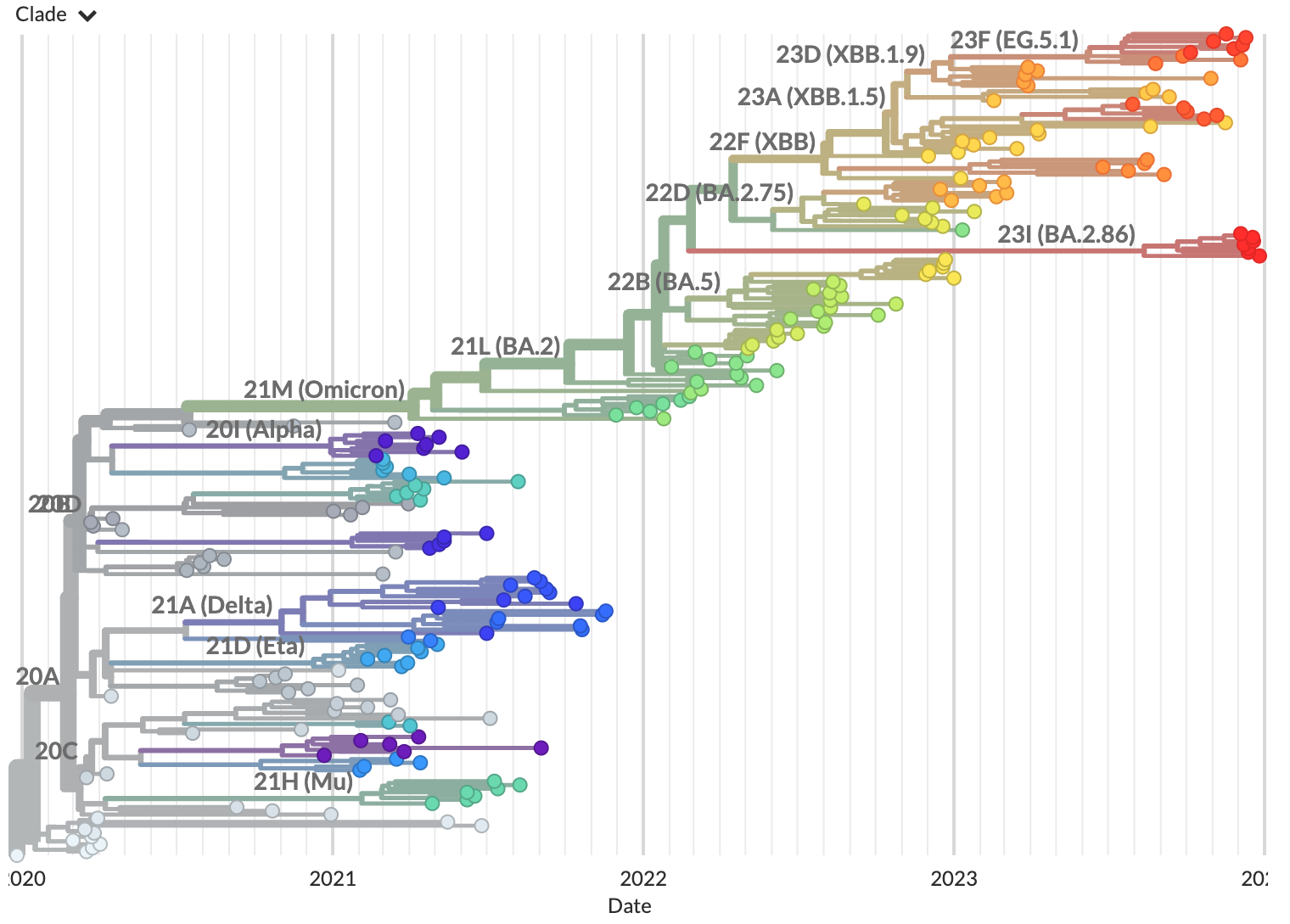
Navigate to http://127.0.0.1:4000/ncov/default-build. The resulting dataset should show a phylogeny of ~200 sequences:
To stop the server, press Control-C on your keyboard.
Note
You can also view the results by dragging the dataset files all at once onto auspice.us:
auspice/ncov_default-build.jsonauspice/ncov_default-build_root-sequence.jsonauspice/ncov_default-build_tip-frequencies.json