Nextstrain overview
Nextstrain has two main parts:
Augur performs the bioinformatic analyses required to produce a tree, map, and other inferences from your input data.
The outputs of Augur form the input for Auspice, which provides the visualizations you see on Nextstrain.org
You can find more information about how these tools fit together here. We’ll come back to Auspice when we get to the visualization section.
First, let’s take a look at how Augur works.
How bioinformatic analyses are managed
At its core, Augur is a collection of Python scripts, each of which handles one step in the bioinformatic analyses necessary for visualization with Auspice.
As you might imagine, keeping track of the input and output files from each step individually can get very confusing, very quickly. So, to manage all of these steps, we use a workflow manager called Snakemake.
Note
There are many other workflow managers out there, such as Nextflow. While we fully encourage you to use whichever workflow tools you prefer, we only provide support and maintenance for Snakemake.
Snakemake is an incredibly powerful workflow manager with many complex features. For our purposes, though, we only need to understand a few things:
Each step in a workflow is called a “rule.” The inputs, outputs, and shell commands for each step/rule are defined in a
.smkfile.Each rule has a number of parameters, which are specified in a ``.yaml`` file.
Each rule produces output (called a “dependency”) which may be used as input to other rules.
Overview of a Nextstrain build
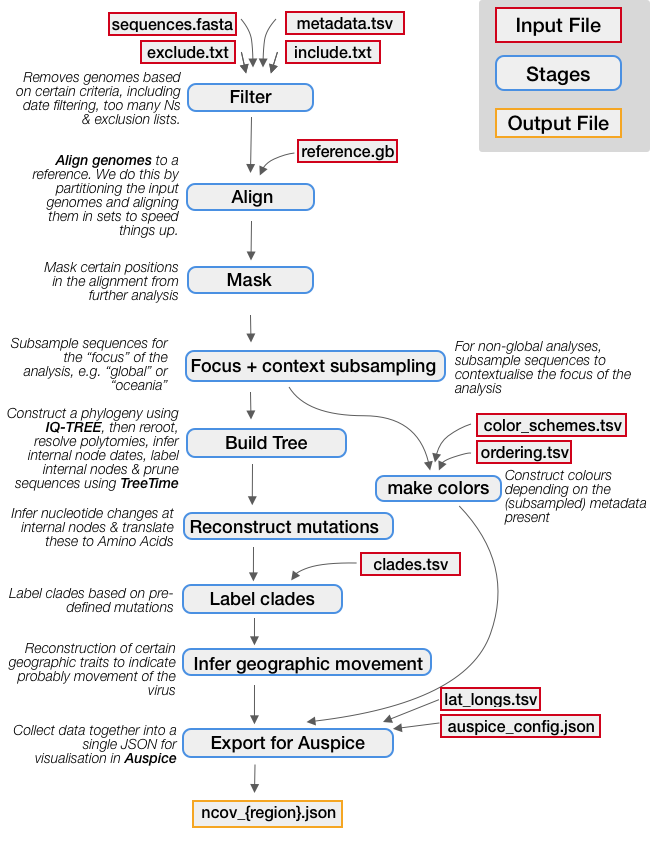
Below is an illustration of each step in a standard Nextstrain build. Dependencies (output files from one step that act as input to the next) are indicated by grey arrows. Input files which must be provided are indicated with red outlines. As you can see in yellow, the final output is a JSON file for visualization in Auspice.
Required input files (e.g. the sequence data generated in the data preparation section, or other files which are part of this repo) are indicated with red outlines. We’ll walk through each of these in detail in the next section.
Running multiple builds
It is common practice to run several related builds. For example, to run one analysis on just your data and another analysis that incorporates background / contextual sequences, you could configure two different builds.
The ncov workflow facilitates this through the builds section in a workflow config file. This is covered in more detail in the genomic surveillance tutorial.
We encourage you to take a look at main_workflow.smk to see what each rule is doing in more detail.
Note
Not all of the rules included are essential, or may even be desirable for your analysis. Your workflow may be able to be made a lot simpler, depending on your goals.