Running a phylogenetic workflow
This tutorial uses the Nextstrain CLI to help you get started running phylogenetic workflows and viewing the datasets you see on nextstrain.org. It assumes you are comfortable using the command line and installing software on your computer. If you need help when following this tutorial, please create a post at discussion.nextstrain.org.
In this tutorial, you will run our example Zika workflow and view the results on your computer. You will have a basic understanding of how to run workflows for other pathogens and a foundation for understanding the Nextstrain ecosystem in more depth.
Prerequisites
Install Nextstrain. These instructions will install all of the software you need to complete this tutorial and others.
Download the example Zika pathogen repository
Pathogen workflows are stored in pathogen repositories (version-controlled folders) to track changes over time. Download the example Zika pathogen repository.
$ git clone https://github.com/nextstrain/zika-tutorial
Cloning into 'zika-tutorial'...
[...more output...]
When it’s done, you’ll have a new directory called zika-tutorial/.
Run the workflow
Phylogenetic workflows use the Augur bioinformatics toolkit to subsample data, align sequences, build a phylogeny, estimate phylogeographic patterns, and save the results in a format suitable for visualization with Auspice.
Run the workflow with the Nextstrain CLI.
$ nextstrain build --cpus 1 zika-tutorial/
Building DAG of jobs...
[...a lot of output...]
This should take just a few minutes to complete. To save time, this tutorial uses example data which is much smaller than our live Zika analysis.
Output files will be in the directories zika-tutorial/data/, zika-tutorial/results/ and zika-tutorial/auspice/.
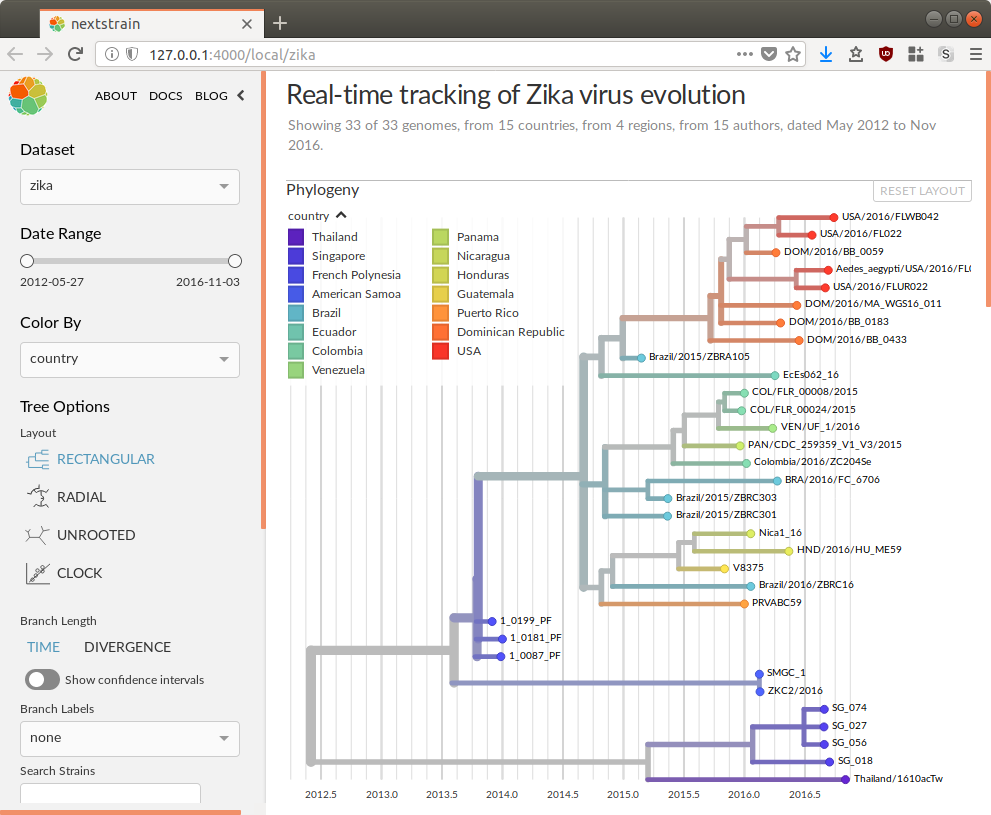
Visualize results
View the resulting phylogenetic dataset using Nextstrain’s visualizations.
$ nextstrain view zika-tutorial/auspice/
——————————————————————————————————————————————————————————————————————————————
The following datasets should be available in a moment:
• http://127.0.0.1:4000/zika
——————————————————————————————————————————————————————————————————————————————
[...more output...]
Open the dataset URL in your web browser.
Next steps
Learn more about the CLI by running
nextstrain --helpandnextstrain <command> --help.Explore the Nextstrain runtime by running ad-hoc commands inside it using
nextstrain shell zika-tutorial/.